Neurofibromatosis tipo 1
La NF1, también llamada enfermedad de Von Recklinghausen, es un trastorno genético frecuente (≈ 1 de cada 2 500 nacidos) que suele diagnosticarse en la infancia por la aparición de manchas café-con-leche, pequeñas pecas en axilas o ingles y, con el tiempo, neurofibromas en piel o nervios. Su cuadro es muy variable: algunas personas solo presentan señales cutáneas leves, mientras que otras desarrollan escoliosis, tumores plexiformes, problemas de aprendizaje o hipertensión.
La alteración se hereda de forma autosómica dominante: si uno de los padres tiene NF1, cada hijo posee un 50 % de posibilidades de nacer con la condición. No obstante, alrededor del 50 % de los casos aparecen “de novo”, sin antecedentes familiares conocidos.
¿Qué ocurre en el ADN?
La NF1 nace de un cambio en un gen situado en el cromosoma 17. Ese gen da lugar a una proteína reguladora del crecimiento celular. Cuando falla, ciertas células —sobre todo las que envuelven los nervios— pueden multiplicarse más de lo debido y formar los tumores característicos. Si te interesa conocer el mecanismo molecular con más detalle, encontrarás una explicación completa AQUÍ.
Aunque hoy no exista cura, el control multidisciplinar y los tratamientos dirigidos (como inhibidores MEK para tumores plexiformes) permiten mejorar la calidad de vida y, en la mayoría de los casos, llevar una vida plena y activa.
Fuente: NF1-EU Registry 2025
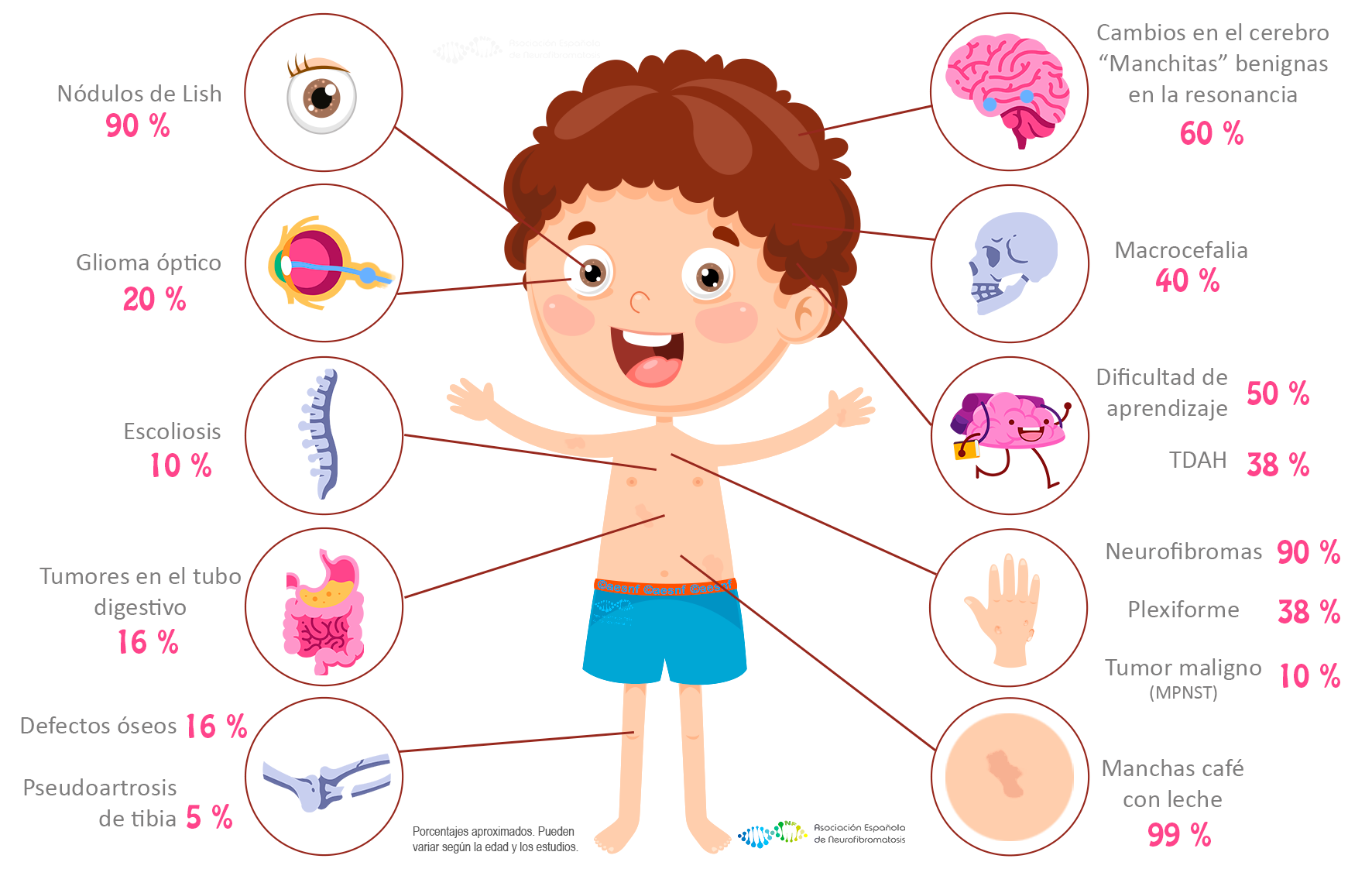
Nódulos de Lisch
Pequeñas máculas pigmentadas en el iris, visibles con lámpara de hendidura a partir de los 5–6 años. No alteran la visión ni requieren tratamiento, pero son muy específicos de NF1 y ayudan a confirmar el diagnóstico cuando otras señales son sutiles.
Glioma óptico
Tumor de bajo grado que afecta al nervio óptico y/o al quiasma. Puede ser asintomático y detectarse en RM rutinaria o manifestarse con disminución de agudeza visual, proptosis (ojo saliente) o pubertad precoz si se extiende al hipotálamo. La mayoría permanece estable y se observa, pero si progresa se tratan con quimioterapia o inhibidores MEK.
Escoliosis
Curvatura lateral de la columna que afecta aproximadamente al 30 % de los niños con NF1. Suele empezar entre los 6 y los 10 años y puede progresar rápidamente. El manejo incluye controles radiológicos, fisioterapia y corsé; las curvas mayores de 40–50° pueden requerir cirugía de artrodesis vertebral para evitar deformidades severas y compromiso respiratorio.
Tumores en el tubo digestivo
La NF1 aumenta el riesgo de GIST (tumor estromal gastrointestinal), sobre todo en intestino delgado, y de feocromocitoma en glándulas suprarrenales. El GIST puede provocar anemia oculta o dolor abdominal; el feocromocitoma cursa con crisis hipertensivas, sudoración y palpitaciones. El diagnóstico suele hacerse con endoscopia, TAC y marcadores bioquímicos; el tratamiento es quirúrgico y, según el caso, con inhibidores de tirosina-quinasa.
Defectos óseos
Alrededor de 1–2 de cada 10 personas con NF1 pueden presentar problemas importantes en los huesos. Los más frecuentes son:
Displasia de la tibia y otros huesos largos: el hueso nace más fino y curvado de lo normal, lo que facilita las fracturas.
Alteraciones en la columna (displasia vertebral, escoliosis): pueden provocar desviación de la espalda y requieren revisión periódica por traumatología.
Displasia del ala esfenoidal: es un defecto en un hueso del cráneo, cerca del ojo, que puede hacer que la órbita se hunda y el ojo parezca más sobresalido.
Todas estas situaciones necesitan seguimiento por traumatología/ortopedia. Sólo una parte de los casos precisan cirugía.
Pseudoartrosis de tibia
Aproximadamente un 5 % de las personas con NF1 presentan pseudoartrosis de tibia.
La pseudoartrosis es una fractura que no llega a soldar bien por sí sola, de modo que el hueso queda inestable. Puede requerir tratamiento con enclavado intramedular, injertos óseos o, en casos complejos, prótesis.
Cambios en el cerebro
En muchas niñas y niños con NF1, la resonancia magnética del cerebro muestra unas pequeñas “manchitas” claras, sobre todo en el cerebelo, tronco cerebral o ganglios basales. Son cambios benignos del tejido nervioso, no son tumores ni se comportan como un cáncer.
Suelen aparecer en la infancia y, en la mayoría de los casos, disminuyen o desaparecen en la adolescencia. Generalmente no producen síntomas y sólo requieren vigilancia en las resonancias de control. Algunos estudios han visto relación entre estas manchitas y las dificultades de aprendizaje, pero esta asociación todavía no está del todo clara.
Macrocefalia
El perímetro craneal rebasa el percentil 97 para la edad en hasta un tercio de los pacientes. Habitualmente se debe a megalencefalia (más tejido cerebral, pero sano) y no produce síntomas. Aun así, se controla con neuroimagen para descartar hidrocefalia u otros procesos que aumenten la presión intracraneal.
Dificultad de aprendizaje
Aproximadamente la mitad de los niños con NF1 presenta alguna dificultad de aprendizaje, como problemas de atención, memoria de trabajo, lectura o cálculo. En torno a un 35–40 % además cumple criterios de TDAH.
La inteligencia global suele ser normal, pero ciertas “funciones ejecutivas” (organizarse, planificar, controlar los impulsos) pueden estar alteradas. Una intervención psicopedagógica temprana (logopedia, apoyo en el aula, técnicas de estudio y ajustes escolares) ayuda mucho a mejorar el rendimiento y la autoestima.
Neurofibromas
Neurofibromas cutáneos y subcutáneos
Son pequeños bultos benignos formados por células de Schwann y tejido conjuntivo que rodean los nervios. Los neurofibromas de la piel y del tejido subcutáneo suelen aparecer a partir de la adolescencia y pueden llegar a ser numerosos (decenas o cientos). La mayoría no duelen ni dan problemas importantes, aunque a veces molestan por roce, picor o por su impacto estético.
Neurofibromas plexiformes
Los neurofibromas plexiformes son tumores más profundos y extendidos, presentes desde el nacimiento, que pueden crecer con el tiempo. Según su localización, pueden comprimir órganos o nervios, producir dolor o causar deformidad visible.
Una pequeña parte de estos tumores puede transformarse en un sarcoma maligno de la vaina nerviosa (MPNST); en conjunto, se estima que alrededor de un 10 % de las personas con NF1 desarrollará un tumor maligno de este tipo a lo largo de su vida.
Cuando la cirugía no es posible o es muy agresiva, fármacos dirigidos como selumetinib o mirdametinib pueden ayudar a reducir el tamaño de algunos plexiformes y mejorar síntomas como el dolor o la limitación funcional.
Manchas café con leche
Son manchas planas de color marrón claro parecidas a “gotas de café con leche”. Aparecen en los primeros meses y van creciendo a la par del niño. Por sí solas no causan molestias, pero tener seis o más (de ≥ 0,5 cm antes de la pubertad o ≥ 1 cm en adultos) constituye uno de los criterios diagnósticos internacionales de NF1. Su presencia temprana facilita la sospecha y el inicio del seguimiento médico. aumentando en número y tamaño con la edad. Son un criterio diagnóstico si se aprecian 6 o más de más de 5 mm en el individuo prepuberal o 15 mm si ha desarrollado ya la pubertad.
Las formas clínicas son muy variadas, con manchas que van desde el marrón claro al oscuro, de color homogéneo, borde suave y redondeado, afectando sobre todo al tronco y las extremidades ya que son raras en la cara, el cuero cabelludo, las palmas y las plantas.
También es muy frecuente (80%) las efélides axilares, estas pecas, que suelen aparecer en la infancia tardía o en la pubertad, pueden afectar a las ingles u otros pliegues pero con mucha menor frecuencia.
Variabilidad de la neurofibromatosis
Manifestaciones clínicas de la NF1 según la edad

De 0 a 2 años
Manchas café-con-leche desde los primeros meses.
Pseudoartrosis o fracturas fáciles (sobre todo en tibia).
Deformidad del ala esfenoidal (rara, pero orientativa).
Glioma del nervio óptico (vigilancia con oftalmología).
Neurofibromas plexiformes congénitos (poco frecuentes a esta edad).

De 2 a 6 años
Pecas en axilas o ingles (signo muy específico).
Nódulos de Lisch en el iris (no afectan la visión).
Glioma óptico u otros tumores del sistema nervioso central.
Retraso del lenguaje o dificultades de aprendizaje iniciales.
Aparición, aún esporádica, de neurofibromas plexiformes.

De 6 a 10 años
Trastorno por déficit de atención y aprendizaje escolar.
Escoliosis o alteraciones óseas progresivas.
Neurofibromas plexiformes con posible crecimiento acelerado.
Cefaleas y, en menor grado, pubertad precoz u otros tumores endocrinos.
Primer aumento del riesgo de tumores no nerviosos (vigilancia oncológica).

Adolescentes
Multiplicación de neurofibromas cutáneos y subcutáneos.
Cambios hormonales que pueden acelerar los plexiformes existentes.
Riesgo (bajo, pero creciente) de transformación maligna de un plexiforme a MPNST.
Hipertensión arterial o vasculopatías asociadas (chequeo anual).
Genes clave de la región cromosómica 17q11.2



REP-A, B, C y D
Son tiras de ADN casi iguales que flanquean al gen NF1. Si la célula las confunde y une la tira “A” con la “C” (o “B” con “D”), corta el ADN en los puntos equivocados y el trozo del medio se pierde (microdeleción) o se duplica (microduplicación).
NF1 es un gen: un tramo concreto de ADN que guarda la “receta” para fabricar la proteína neurofibromina.
La neurofibromina es como un interruptor: cuando está encendida, desactiva la vía RAS – una señal que impulsa a las células a crecer y dividirse.
Situación normal
Poseemos dos cromosomas 17 (uno de cada progenitor), y en cada uno hay una copia funcional de NF1. Con ambas copias activas, la neurofibromina mantiene a raya la vía RAS y las células crecen de forma controlada.
Mutaciones o pequeños añadidos/pérdidas de letras dentro del gen NF1
A veces se sustituyen, se añaden o se pierden unas pocas “letras” de la receta. El gen sigue en su lugar, pero una de las copias deja de funcionar. Otras veces se borran o se repiten fragmentos algo más grandes —uno o varios “párrafos” del gen— sin afectar a los genes vecinos. Esa copia tampoco produce neurofibromina útil.
En ambos supuestos sucede lo mismo: solo queda operativa una de las dos copias del gen NF1, la cantidad total de neurofibromina se reduce a la mitad y aparecen los signos de la NF1 clásica. ( ≈ 88–93 % de los casos de NF1).
Microdeleción 17q11.2 (−)
El recuadro punteado muestra el tramo que se pierde.
La célula, al intentar recombinar, confunde REP-A con REP-C (porque se parecen mucho). Corta en A, vuelve a enganchar en C y todo lo intermedio desaparece → queda una sola copia funcional de NF1. ( ≈ 5–11 % de los casos de NF1).
Microduplicación 17q11.2 (+)
El bloque amarillo doble indica que el mismo tramo se copió dos veces. NF1 queda triplicado (3 copias). El freno no falta, pero la dosis de varios genes es excesiva → aparece un síndrome distinto (no desencadena NF1).



¿Qué es RAS?
RAS es una proteína-interruptor, como un botón de encendido del crecimiento celular.
- RAS + GTP → el botón está “ON” y la célula recibe la orden de crecer y dividirse.
- RAS + GDP → el botón pasa a “OFF” y la señal de crecimiento se apaga.
GTP y GDP son, por así decirlo, los “cartuchos de energía” que marcan si RAS está encendido o apagado.
La neurofibromina (fabricada a partir del gen NF1) acelera el cambio de GTP a GDP; es decir, apaga RAS a tiempo. Cuando falta neurofibromina, RAS se queda atascado en “ON” y eso desencadena los problemas típicos de la NF1.
Medicamentos dirigidos a la vía RAS–MAPK
Bloquear directamente a RAS es casi imposible: la proteína carece de un “hueco” donde encaje un fármaco. Dos pasos más abajo se encuentra MEK-1/2, que sí tiene un bolsillo aprovechable; al inhibir MEK se interrumpe la señal que proviene de RAS antes de que active los genes de crecimiento.
- RAS enciende RAF.
- RAF activa MEK.
- MEK fosforila a MAPK—también llamada ERK—que entra en el núcleo y pone en marcha genes de crecimiento y división celular.
Por eso se habla de la “vía RAS-MAPK”: empieza en RAS y termina en MAPK/ERK.
Los medicamentos como selumetinib y mirdametinib se enganchan a MEK, cortando la señal antes de que llegue a MAPK y, así, frenan la proliferación de las células afectadas.