¿Qué es la Neurofibromatosis?
La neurofibromatosis (NF) es un conjunto de trastornos genéticos poco frecuentes que afectan principalmente al sistema nervioso. Su origen está en mutaciones del ADN que alteran el crecimiento normal de las células, dando lugar a tumores en los nervios, generalmente benignos, pero que pueden provocar complicaciones médicas importantes.

Neurofibroma plexiforme, es un tipo de tumor de la vaina nerviosa
Aunque los primeros casos clínicos se describieron en el siglo XIX, no fue hasta finales del siglo XX cuando se identificaron los genes responsables y se clasificaron correctamente los distintos tipos:
NF tipo 1 (NF1)
NF tipo 2 relacionada con schwannomatosis (NF2-SWN)
Schwannomatosis (SWN)
Cada una tiene características distintas, pero todas comparten un impacto importante sobre la salud y la calidad de vida.
¿A cuántas personas afecta?
La neurofibromatosis es considerada una enfermedad rara, pero no es tan infrecuente como podría parecer:
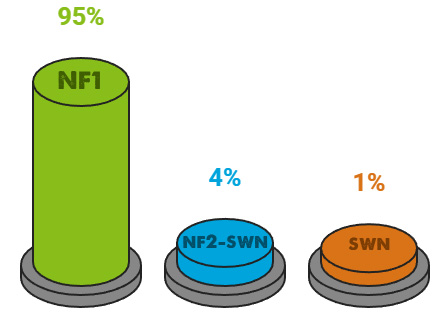
En torno a 1 de cada 2.500 nacimientos presenta NF1, lo que significa que cada año nacen unos 2.500 niños con esta enfermedad solo en España.
Los otros subtipos son menos frecuentes, pero también están presentes en cientos de familias.
¿Cómo se hereda?
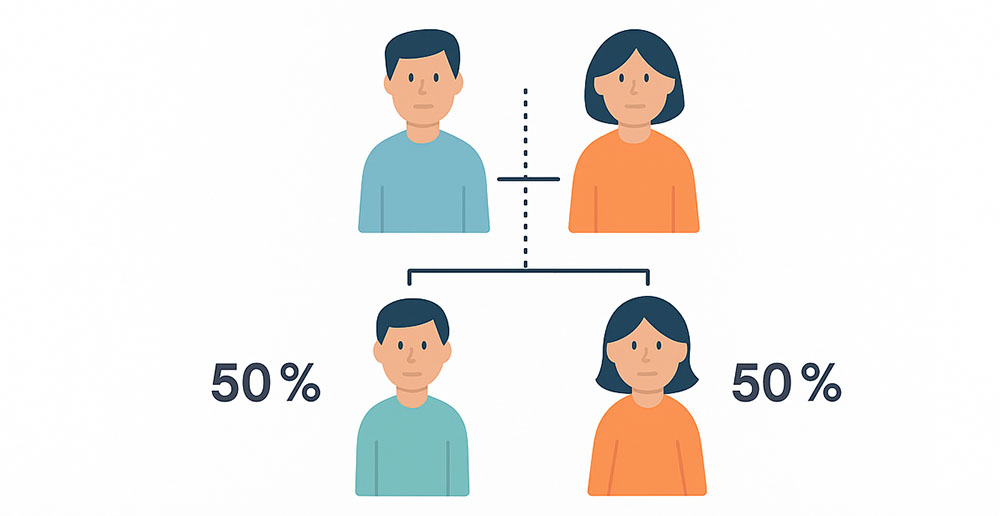
La NF puede aparecer de forma hereditaria (cuando uno de los progenitores transmite el gen mutado) o como una mutación espontánea, sin antecedentes familiares previos.
En ambos casos, el riesgo de transmitirla a los hijos es del 50 % si uno de los padres está afectado. Por eso el asesoramiento genético es tan importante.
Más allá del diagnóstico
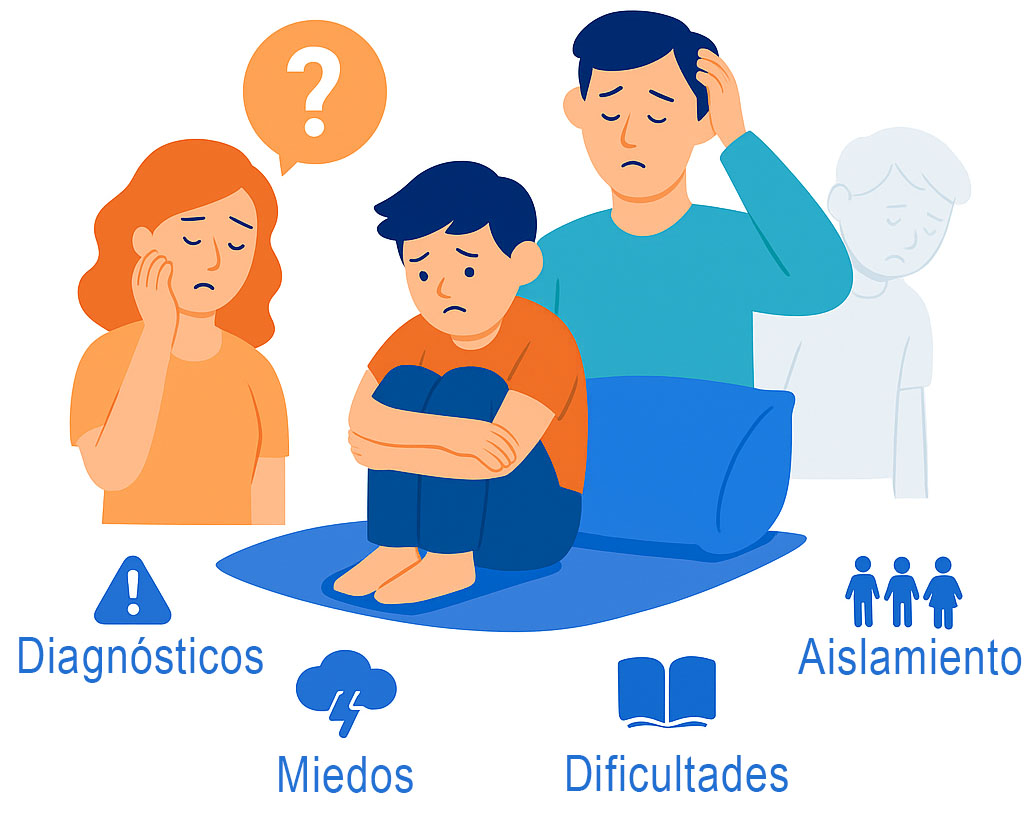
La neurofibromatosis no solo afecta al cuerpo. También puede tener un fuerte impacto emocional y social, tanto en el afectado como en su familia:
Diagnósticos inciertos y retrasados
Miedos constantes a nuevas complicaciones
Aislamiento o incomprensión social
Dificultades escolares o laborales
En AESNF trabajamos para acompañar, informar y conectar a todas las personas que viven con esta enfermedad. Porque conocer la NF es el primer paso para no sentirte solo frente a ella.


